
In March of 2019, for the second year in a row, I attended the Bay Area Society for Neuroscience’s biggest event of the year, SfN Wonder. The event featured five distinct speakers—Dr. Brad Zuchero, Yung Leong, Dr. Blair Kaneshiro, Teresa Bell-Stephens, and Cole Holderman—who each talked about a different aspect of neuroscience that they specialized in. All of the speakers had amazing presentations, but I found Dr. Zuchero, Ms. Bell-Stephen’s, and Mr. Holderman’s talks especially insightful and relevant to my own interests. Dr. Zuchero spoke on neurophysiology and glial cells, Ms. Bell-Stephens spoke about media in neuroscience in the context of Moyamoya disease, and Mr. Holderman spoke on health awareness with regards to Huntington’s.
Dr. Brad Zuchero started his lab at Stanford 2 years ago with a special focus on glial cells. He told us that more than half of our brain cells are glial cells, yet traditionally people did not study them; they were dismissed as just the “glue” that holds everything together. In this talk, Dr. Zuchero challenged that notion by telling us about the work he and his late mentor Dr. Ben Barres have done. What was first necessary to study the role and function of glia was to isolate them from the neurons and then divide the glia into their various types, the important ones being microglia, OPCs, oligodendrocytes, and astrocytes. Dr. Barres figured out a technique known as immunostaining to purify and culture neurons and glia: by coating antibodies on various Petri dishes and passing a dissociated brain on each, specific antigens bind to the desired constituent part, separating it from the whole. Therefore, he got a petri dish of neurons, a petri dish of astrocytes, a petri dish of microglia, etc.
Dr. Zuchero then focused on the role of astrocytes, microglia, and oligodendrocytes. Astrocytes are completely spread throughout the brain; a single human astrocyte can ensheath 400,000 synapses. Researchers have seen that if retinal ganglion cells, a type of astrocytes, are purified away from the neurons, the neurons do not form synapses. What is perhaps even more interesting is that astrocytes do not need to touch the neuron; they just need to be nearby to release soluble factors that can diffuse to form and strengthen synapses. Like astrocytes, microglia extend a great deal throughout the central nervous system, serving as innate immune cells, sensing and responding to pathogens or injury. As seen in a mouse model, during development, microglia eat synapses in order to prune the excess neurons so that we only have the strongest and most proper ones in place. As Alzheimer’s sees massive synapse loss, it is possible that microglia overeating synapses could be the cause or at least contributing to this neurodegenerative disease. Indeed, the genes that have been found to be risk factors for Alzheimer’s often are expressed by glial, not neuronal, cells, yet almost every drug for the neurodegenerative disease has targeted neurons, not glia.
Dr. Zuchero’s lab is specifically focused on myelin and the oligodendrocytes that make the myelin sheath, extending and spiraling tight around the axons. Axons are often coated in myelin as the lipid-rich material acts as an electrical insulator, helping action potentials flow fast through it. It has been shown that stimulation of neurons using optogenetics, using light to control activity, for example, can cause adaptive myelination. You get rapid induction of oligodendrocyte formation as well as the creation of new myelin. Thus, neuronal activity not only forms and strengthens new synapses but promotes changes and remodeling of myelin.
The big takeaway from Dr. Zuchero’s presentation was that glia is paramount to how we look at nervous system disorders. Stroke, for example, sees the massive death of neurons, but it also sees the death of other cells like astrocytes. As mentioned previously, without astrocytes, neurons cannot form synapses and thus die; however, nearly every drug trial to date for stroke has targeted the neurons instead of the glia. Glia is the untapped therapeutic target for brain disorders, and they deserve much more attention than they currently get.
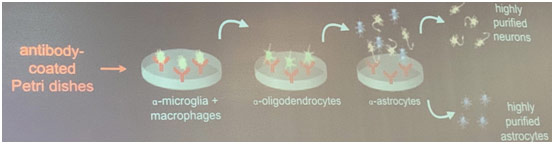
Immunopanning Method
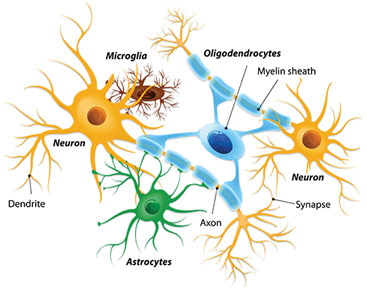
Neurons and Neuroglial Cells
Ms. Bell-Stephens has been a registered nurse for 40 years now. She used to be a critical care nurse, enjoying the adrenaline rush of working in the ICU, before she shifted to working in neurosurgery. She spoke on how media interacted with Moyamoya disease to increase awareness about the disease. Moyamoya is a rare disease characterized by the progressive bilateral occlusion of the internal carotid arteries, causing them to shrivel up, leading to no blood to enter the ACA (anterior cerebral artery) nor the MCA (middle cerebral artery). “Moyamoya” in Japanese means “puff of smoke” because patients develop small compensatory fragile arteries that look hazy on cerebral angiography. The disease affects about 0.086/100,000 people in the US, but is much higher for Asians with a prevalence of 6/100,000. The disease also mostly presents between 5-7 or 35-45. That being said, Moyamoya can affect all races and ages. In fact, a majority of the patients who come to Stanford for treatment for Moyamoya disease are Caucasian, which just highlights the importance of not labeling Moyamoya as an “Asian disease.” Moyamoya can be so devastating because the fragile collaterals cannot supply the brain with an adequate blood supply and thus adequate oxygen. Therefore, patients have TIAs (mini-strokes), strokes, hemorrhages, seizures, and headaches. In adult patients, the symptoms present in 55%, 59%, 16%, 14%, and 49% of patients respectively. In pediatric patients, it is 39%, 42%, 6%, 11%, and 23% respectively. Ms. Bell-Stephens told us that the children’s symptom percentages are likely lower than they actually are because young children have a tendency to not tell anyone about their symptoms because they do not understand the significance of the “funny feeling” in their hand for example.
A Washington University in St. Louis study found that even with the best medications, 65% of patients with Moyamoya disease had a stroke after 5 years. Based on that dismal statistic, the healthcare team at Stanford is striving to combine pharmacotherapy with surgery to get the best outcomes for their patients. Before they do the surgery, they put the patient on midodrine and Florinef to raise the patient’s blood pressure so that the brain can get enough blood while giving the patient the blood thinner aspirin to prevent strokes. They then try to do direct revascularization surgery. Generally, the more symptomatic side, or the nondominant side in a nonsymptomatic patient, is revascularized first while the other side is revascularized after a week. One part of a scalp artery, more specifically the STA (superficial temporal artery), is then connected to the MCA in an end-side anastomosis in order to improve blood flow to the brain. Unfortunately, this kind of STA-MCA bypass is the only surgery possible as the internal carotid arteries affected by Moyamoya arteries cannot be repaired.
DJ, an Air Force sergeant from Kansas afflicted by Moyamoya disease, could not find a surgeon who could treat him. Because Moyamoya is such a rare disease, DJ had a hard time getting diagnosed, and after getting diagnosed, no one nor search engine seemed to be able to offer him any information of value. Thus, as a patient, he did a literature search to read up on the disorder, becoming a self-advocate. He came for treatment after discovering that Stanford has a Moyamoya Center and proceeded to create https://moyamoya.com to help those who want to learn more about this rare disorder but have been disappointed with most resources available to them. Stanford has done 1700 revascularization procedures to date, but they had an enormous bump in the number of cases they received in 2004 and now have a sustained surgical population because this patient advocated for himself on the internet. Another patient dedicated May 6th as World Moyamoya Day, creating a Facebook page and getting multiple states to officially recognize the day in order to bring awareness to this unusual disease. With patients and Stanford putting the word out and bringing awareness through social media, more and more patients have been able to obtain treatment for this disorder and clear their misconceptions, like the notion that one is not able to get pregnant if they have Moyamoya. Ms. Bell-Stephens showed us that the impact of media on neuroscience can be enormous; in this case, it worked to improve the quality of life and outcomes with patients with Moyamoya disease.
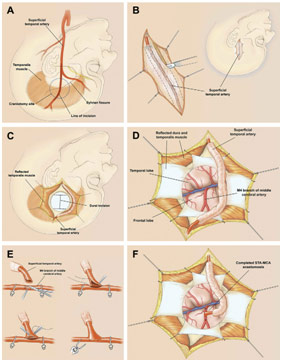
Revascularization Surgery with STA-MCA Anastomosis
Mr. Cole Holderman is a senior at Stanford University majoring in Human Biology, and he is also the student director of the Huntington’s Outreach Project for Education at Stanford (HOPES). He spoke on health literacy with regards to Huntington’s Disease (HD). HD is a rare dominant neurodegenerative genetic disorder. Generally, it tends to occur in 5-7 in 100,000 people and can be passed down with only one copy of the mutant gene; therefore, a parent with the disorder has a 50% chance of passing it down to their child. Specifically, the issue is with the Huntingtin (HTT) gene where there is a small region known as the CAG repeat area. 8-29 repeats mean the patient has no risk of developing the disorder (17 is average), 29-35 repeats means the patient has a premutation, and 36-40 means the patient has reduced penetrance (can develop symptoms and pass it on). You get adult-onset Huntington’s with more than 40 repeats and juvenile-onset Huntington’s (10% of HD patients) with more than 60 repeats. The excess CAG repeats cause a protein with an excess of the amino acid glutamine to form, which is toxic to neurons and causes atrophy in the basal ganglia of the brain. Common symptoms of this constant and progressive degeneration of the nervous system include chorea (inability to stop oneself from moving), behavior changes (inability to stop the behavior from happening), and paralysis, among others. Because it is such a rare disorder, HD faces issues in diagnosis, access to treatment, and funding for research. Research is so paramount for HD because there currently exists no cure and the therapies approved for the disorder just treat the symptoms.
In 1967, patients, clinicians, and families formed the Huntington’s Disease Society of America to advocate for policies that benefit those suffering from this rare disease. In 2000, Huntington’s Disease Lighthouse was created as an online forum for patient families and to announce new research updates. However, Professor Bill Durham realized that something was missing when a family with Huntington’s told him that they had no idea what the disorder is, why one of their members has it, and what the treatment looks like. Professor Durham wrote that “you could see that no one was focused on the science behind HD.” Most of the resources explaining the disorder were geared toward health care professionals, not their families. The patients had no knowledge of their condition and next steps, so they could not form a new normal within the context of their condition. He decided to provide some source that could show what Huntington’s is at the reader’s unique level. HOPES was thus founded and had great success in making patients community experts in the disease, becoming one of the leading HD information sources in the world with 1 million readers in 2007. Now, HOPES has begun to interface with local support groups and organizations while also providing information on research, clinical trials, HD in media, and laws affecting HD patients. With other organizations like HDYO and HDBuzz helping raise awareness and initiatives like the HD Parity Act to put HD into a category of disorders that get federal disability relief, Mr. Holderman told us that the future of HD is looking a lot brighter than it was years ago when nearly no one even knew what the disorder is. There is even a very promising Phase II clinical trial for a potential disease limiting treatment that uses ASOs (allele-specific antisense oligonucleotides), which are little strands of mRNA that bind to other mRNAs. The hope is to silence the Huntingtin gene to stop it from making the atrophy-causing proteins in order to extend the lives of patients with HD.
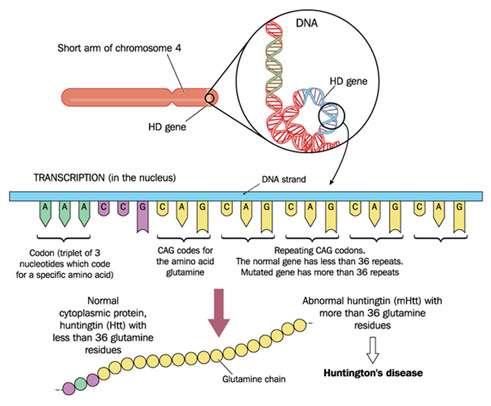
Huntingtin Gene CAG Repeats
The experience of going to SfN Wonder for my second year now was so great because I got to meet and hear from professionals who are experts in their fields.
REFERENCES
Brookshire, Bethany. Neurons and Neuroglial Cells. 16 Oct. 2017. Science News for Students, www.sciencenewsforstudents.org/blog/scientists-say/scientists-say-glia. Accessed 3 Apr. 2019.
Guzman, Raphael, et al. “Clinical Outcome after 450 Revascularization Procedures for Moyamoya Disease.” Jounral of Neurosurgery, vol. 111, no. 5, Nov. 2009, pp. 927-35, stanfordhealthcare.org/content/dam/SHC/clinics/moyamoya-center/docs/pdf-moyamoyadiseaseclinicaloutcome.pdf. Accessed 3 Apr. 2019.
Guzman, Raphael, and Gary Steinberg. Revascularization Surgery with STA-MCA Anastomosis. Semantic Scholar, www.semanticscholar.org/paper/Direct-bypass-techniques-for-the-treatment-of-Guzman-Steinberg/2b316c92feb4dc274ec30224e45b0a93436ad38e. Accessed 3 Apr. 2019.
The HTT Mutation. NIH, ghr.nlm.nih.gov/condition/huntington-disease. Accessed 3 Apr. 2019.
“Overview of Huntington’s Disease.” Huntington’s Disease Society of America, hdsa.org/what-is-hd/overview-of-huntingtons-disease/. Accessed 3 Apr. 2019.








{kind=link}
Sounds actually interesting. Is it ok to volunteer one or two questions?