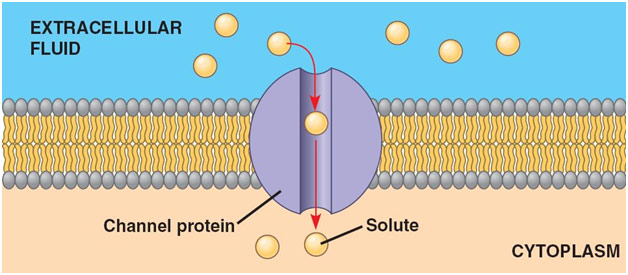
Cystic fibrosis affects greater than 30,000 people in the US with patients having an average life expectancy of 37.5 years, making this genetic disease quite devastating to those afflicted with it. “Cyst” refers to fluid-filled pockets, and “fibrosis” refers to the formation of excess connective tissue, both of which tend to happen in the pancreas of patients with cystic fibrosis. Ultimately though, cystic fibrosis is a disease that deals with diffusion and osmosis. All cells have a membrane made of a phospholipid bilayer with outward-facing hydrophilic heads and inward-facing hydrophobic tails designed to separate the contents of the cell from the contents of the environment. Ions cannot pass through the cell membrane through simple diffusion, so they typically must go through channel proteins. In cystic fibrosis, issues with the CFTR (cystic fibrosis transmembrane regulator) channel protein arise, leading to ion imbalances that have severe consequences on the entire body.
Specifically, a genetic mutation in the CFTR gene is the cause of the channel protein issues. The mutations can be put into three groups. Class I and II mutations are characterized by the CFTR protein never making it to the cell membrane because the RNA is faulty or the protein is folded incorrectly, respectively. Class III and IV mutations are characterized by a channel protein that does not function properly and thus cannot serve its purpose. Class V and VI mutations see there not being enough CFTR channel protein because either enough is not made or the channel lacks stability, respectively. Nearly 90% of cystic fibrosis patients have a Class II mutation with the most common being the ∆F508 mutation where the amino acid phenylalanine is improperly deleted. As cystic fibrosis is an autosomal recessive condition, if both parents are carriers and thus unsymptomatic for the disease, it can be passed down to their child in one-fourth of cases.
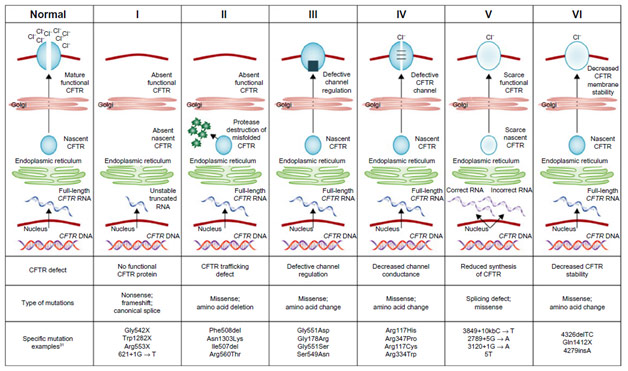
Normal CFTR Protein vs. Different Classes of Mutations
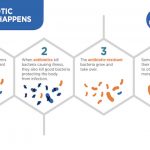
CFTR is such a fundamentally important channel protein in the lungs because it transports chloride ions out into the mucous secretions. Typically, the increased concentration of chloride ions in these secretions leads them to pull in water through osmosis, hydrating and thinning these secretions out. This is important because the mucus is designed to trap bacteria that cilia (tiny hairs) subsequently push out the body. Without these chloride ions, however, the secretions become quite viscous, which leads to some airways being clogged altogether. Furthermore, because of the thick, sticky mucus, the cilia cannot move the bacteria out of the lungs. The stagnant mucus thus becomes a breeding ground for inhaled bacteria, leading the patient to develop chronic severe respiratory infections in what is called cystic fibrosis exacerbation. While antibiotics can treat these infections, the repeated use for patients with cystic fibrosis tends to make the bacteria antibiotic-resistant, which can prove near impossible to treat. Eventually, due in large part to damage to the respiratory tract, the patient can develop respiratory failure, which is often fatal without a lung transplant.
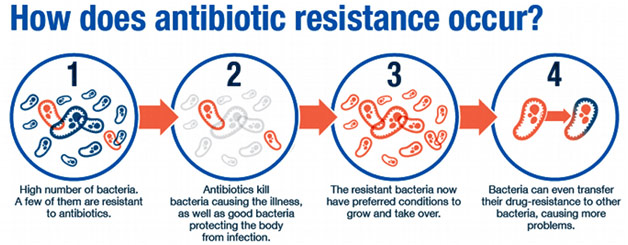
Mechanism of the Development of Antibiotic Resistance
While lung involvement is most characteristic of cystic fibrosis, the disease also often affects other parts of the body such as the pancreas and intestines. For similar reasons as in the lung, secretions in the pancreas become dehydrated and thickened, leading them to block the pancreatic ducts, which deliver vital digestive enzymes to the small intestine. Without these enzymes, the patient does not efficiently absorb protein and fat from their foodl; this is known as pancreatic insufficiency. When undigested food is combined with yet more thick mucus in the intestines, the patient can develop distal intestinal obstruction syndrome where there is a blockage in the small intestine. The obstruction is generally flushed out with laxatives or an enema but, sometimes, with surgery. This complication happens in about one-tenth to one-fourth of cystic fibrosis patients. One of the earliest diagnostic indications of cystic fibrosis is actually meconium ileus, which is when the infant’s first stool causes a similar type of blockage in the first part of their small intestine.
Blockages in the pancreatic ducts cause more complications than poor digestion though. In fact, the pancreatic enzymes, holed up in the pancreatic ducts, can begin to degrade the pancreas and cause pancreatitis, a severe case of inflammation of the organ. Eventually, the enzymes can even compromise the endocrine function of the pancreas, leading the patient to develop Type I diabetes because the glucose-producing beta cells are destroyed. Cystic-fibrosis-related Type-1 diabetes occurs in 40% of these patients above the age of 30.
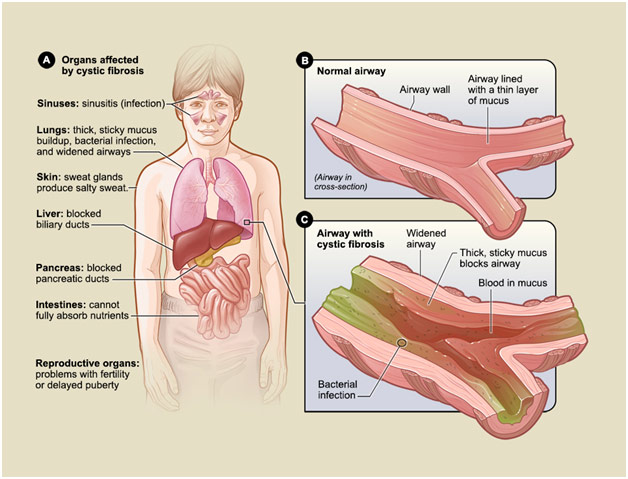
Organs Affected by Cystic Fibrosis
In the US, all newborns undergo testing for cystic fibrosis where blood obtained from a heel-prick is tested for immunoreactive trypsinogen (IRT), an enzyme indicative of pancreatic damage. In presence of high IRT, another testing may be used to help confirm the cystic fibrosis diagnosis. However, a significant portion of the population was not tested for cystic fibrosis as newborns because of the relative newness of this policy. A genetic test may be done to detect the presence of a mutation in the CFTR gene; however, a sweat chloride test is a standard to diagnose cystic fibrosis. A sweat chloride test is generally performed by using chemicals to stimulate the patient’s sweat glands. The sweat is then collected and tested for its chloride content with a result higher than 60 mmol/L indicating a cystic fibrosis diagnosis.
Cystic fibrosis treatment usually falls into one of a few categories: airway clearance therapy, mucolytics, nutrition-based therapy, anti-inflammatories/bronchodilators, and therapies to reverse the underlying abnormalities. Airway clearance therapy, usually achieved through manual chest physiotherapy or a Vest device, seeks to loosen the thick mucus and make it easier to cough out. Mucolytics, such as hypertonic saline or dornase alfa, seek to help the patient thin out their mucus and thus more easily remove it. Nutrition based therapy is focused on supplementing the patient with extra calories and fat-soluble vitamins to make up for their impaired digestive system as well as on giving them oral pancreatic enzymes to help them better absorb nutrients from their food. Anti-inflammatories and bronchodilators, such as symedko and albuterol respectively, seek to help keep the patient’s airways open so that they may more easily breathe. Lastly, therapies that seek to reverse the underlying abnormalities are quite promising. Orkambi is a combination drug of lumacaftor and ivacaftor, which works in patients with the ∆F508 mutation CFTR mutation by helping the protein properly fold and then ensuring that even the improperly folded channels still stay open so that chloride ions may pass through.
Airway Clearance Vest Therapy
The recent development of Orkambi to treat the underlying cause of cystic fibrosis rather than its symptom proves quite promising and indicative that greater awareness and research into cystic fibrosis provides tangible results. Much work is currently being done to create a gene therapy that would replace the defective CFTR gene with a properly functioning one, which would hopefully cure the patient of this devastating disease. Survival for patients with cystic fibrosis has doubled in the past half-century, so let us hope that one day, with more research and attention, patients with cystic fibrosis may live fully without any fear of their disease.






{kind=link}